Most treatments targeting Alzheimer’s disease focus, to no avail, on directly reducing the accumulation of cognitive function-impairing amyloid plaques in the brain. A research team from EPFL, working in animal models and human neuronal cells, has shown that tackling mitochondrial defects might instead be the key to designing new efficient treatments (Nature 552 187).
A hallmark of Alzheimer’s disease is the accumulation of toxic plaques formed by the abnormal aggregation of beta-amyloid protein inside neurons. While no definitive cure exists, many treatments have tried to reduce the development of those plaques. Recently, a tentative vaccine managed to bind virus-like particles to them, opening the window for a potential immunogenic therapy for Alzheimer’s disease (Towards a vaccine for Alzheimer’s disease).
However, none of these approaches have been validated in humans or proved to be efficient. Consequently, other methods are being investigated, and Johan Auwerx and his team at EPFL are taking an original path: approaching Alzheimer’s as a metabolic disease.
Under this paradigm, Alzheimer’s disease would be caused by defects in the chemical reactions of the body’s cells, which would alter their normal metabolism, such as the process of converting food to energy on a cellular level.
To support this view, the researchers used the fact that mitochondria, the energy-producing powerhouses of cells, are dysfunctional in the brains of Alzheimer’s patients. As cells age, they are exposed to increased levels of damage, which affect their mitochondria, making them dysfunctional. Such mitochondria would normally be replaced by autophagy, but over time, the cells become less efficient at removing defective mitochondria. They cannot defend themselves against Alzheimer’s disease.
A problem of quality control?
The researchers identified two key mechanisms controlling the quality of mitochondria: mitophagy, a process that recycles defective mitochondria; and mitochondrial unfolded protein response (UPRmt), which protects mitochondria from stress stimuli. These actions are key to delaying or preventing excessive mitochondrial damage. The team therefore hypothesized that boosting the activity of these two mechanisms might help to slow the progression of the disease.
To test their hypothesis, the researchers attempted to turn on the UPRmt and mitophagy processes by administering various in vitro and in vivo organisms with two commonly-used drugs: the antibiotic doxycycline and the vitamin nicotinamide riboside.
Next step: human trials
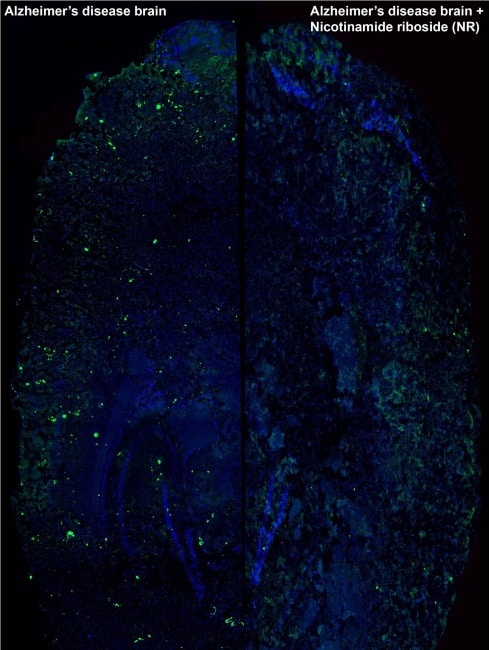
In a worm model of amyloid-beta disease, treated animals showed a remarkable improvement in health, performance and lifespan when compared with the control group. Protein analysis also displayed a significant reduction of plaque formation in worms exposed to the drugs. The same results were observed in cultured human neuronal cells.
Experiments in a mouse model of Alzheimer’s disease were even more encouraging. Not only did the mice exhibit the same improvement in their mitochondrial function and a reduction in the number of amyloid plaques, but the researchers also observed a striking normalization of their cognitive function, the main objective for any treatment of Alzheimer’s disease.
According to the World Health Organization, Alzheimer’s disease affects 35 million people around the world, with the figure expected to exceed 100 million by 2050. With life expectancy increasing, finding effective approaches to tackle Alzheimer’s is a necessity. The results of Auwerx’s work are yet to be validated in humans, but they may pave the way for a metabolic response to the disease. The fact that the two mitochondrial quality control processes identified can be activated in the same way in worms, mice and cultured human cells is cause for optimism.