Small angle X-ray scattering (SAXS) is one of a number of biophysical techniques used for determining the structural characteristics of biomolecules. Daniel Franke and colleagues from the European Molecular Biology Laboratory have recently published a machine learning-based method to classify biomolecules using existing SAXS data (Biophys. J. 114 2485).
The method can be used to classify shape, as well as estimate structural parameters such as the maximal diameter or molecular mass of the molecule under study. These estimates may then serve as a valuable method for validating expected values.
The team decided on a set of shape classifications for biomolecules: compact spheres, flat discs, extended rods, compact-hollow cylinders, hollow spheres and flat rings. They used simulations to obtain idealized scattering profiles of each of these different geometries across a range of heights, widths and lengths ranging from 10 to 500 Å.
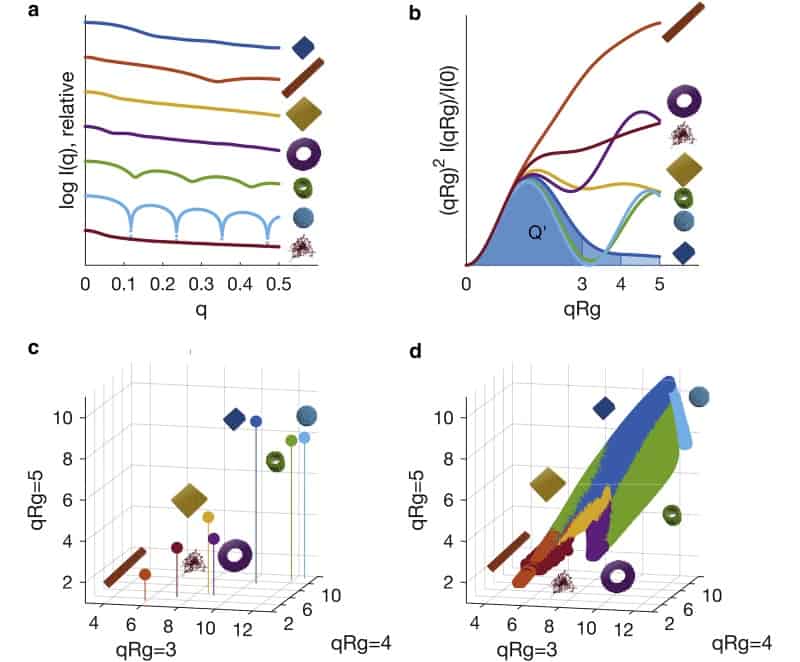
The researchers used innovative data reduction approaches to reduce each of the scattering profiles to a point in normalized apparent volume space, V. Representing the data in this way is advantageous because structures that share similar structural characteristics will occupy a similar position in V space.
The process of classifying an unknown scattering profile then amounts to calculating its position in V space and locating the nearest points in V space for which parameters are already known. The new parameters can then be estimated by taking a weighted average of these “nearest neighbour” points in V space. A machine can be programmed to perform all of these steps.
Using machine learning
The team simulated some 488,000 scattering patterns and used these to train an algorithm to categorize different scattering patterns. Each scattering pattern was then removed in turn, and the remaining data used to predict the shape classification of the removed pattern.
This training procedure allowed the researchers to refine the weights assigned to the nearest neighbour structures in V space, so as to maximize the accuracy of the machine classification.
Predicting structural parameters
To test the predictive power of the shape classification method, the researchers harvested scattering data from the Protein Data Bank (PDB) and the Small Angle Scattering Biological Data Bank (SASBDB).
From the atomic structures stored in the PDB, they used CRYSOL software to generate scattering intensities, as well as values of structural parameters such as the maximal diameter and molecular mass. After mapping the known structures to V space, an equivalent algorithm was then used to predict the structural parameters based on the generated scattering intensity. Here, the machine prediction was within 10% of the expected value in 90% of cases.
The SASBDB provides scattering intensity as well as user generated values of structural parameters such as the maximal diameter. The researchers also observed good agreement from the structures collected from the SASBDB, with the machine predicting a small, systematically lower value for the maximal diameter. This offset reflects the fact that molecules tend to occupy an extended configuration in solution.
The protocol developed by the team shows that data mining has significant potential to increase the efficiency and reliability of scattering data, which could have huge benefit for the biophysics community.