Recent developments in X-ray crystallography at synchrotron radiation sources and progress in the production of good-quality protein crystals are leading to important advances in our knowledge of protein structure and function. Naomi Chayen and John Helliwell describe how recent advances in protein crystallography are providing new insights into their structure and properties.

It is impossible to overstate the importance of proteins to plant and animal life. Much of the tissue in the human body is made of protein, as are all of the enzymes that catalyse reactions in the body, the globins that transport and store oxygen, the antibodies responsible for the body’s immune response and hormones such as insulin.
Proteins are macromolecules made from combinations of the 20 naturally occurring amino acids. A typical protein contains about 300 amino acids, although some proteins can contain as many as 1000, and multi-macromolecular protein and nucleic-acid complexes represent further levels of complexity. Amino acids are small molecules that are made up mostly of carbon, hydrogen, nitrogen and oxygen, although two amino acids also contain sulphur.
The order of the amino acids in a protein is determined by the sequence of “base pairs” in the deoxyribonucleic acid (DNA) that is found inside every living cell. In humans this DNA is divided among 23 pairs of chromosomes that collectively make up what is known as the human genome. A gene is the sequence of base pairs that carries the code for a single protein and there are about 100,000 genes in the human genome. Scientists from all over the world have joined forces to determine the order of the base pairs in the human genome and physics-based techniques are playing their part in that effort (see “Physics and the Human Genome Project” by Norman Dovichi in Physics World September 1997).
Just as important as chemical composition, however, is the shape or conformation of the protein because this determines its detailed chemical and biological function – be that enzyme catalysis, viral infection, oxygen transport or the immune response. The shape is determined by the orientation of each amino acid relative to its two neighbours, with the amino acids linking in a way that is energetically “comfortable”. Although there are only three or so sterically acceptable orientations for each pair of amino acids, the total number of shapes possible for a typical protein containing 300 amino acids is thereby huge, about 3300.
Protein challenges
One of the great mysteries of biology is how a long chain of amino acids “folds” into its final working (i.e. active) shape within seconds – if the protein followed a random walk to its final structure, it would take much longer. This is a problem that has attracted the attention of many physicists as it has parallels with systems studied in statistical mechanics.
Progress has been made in terms of the secondary structures within a protein: two commonly found substructures are the so-called alpha helices and beta sheets. However, large parts of the structure cannot be predicted. Moreover, even for those substructures like helices that can be predicted, the precise placement of the atoms or chemical groups that are central to the protein’s function cannot. These side chains determine the reactivity and intermolecular “recognition” of the proteins.
There is therefore great demand for fast and efficient techniques to determine the 3-D structures of proteins in terms of bond distances and angles. Many physics-based techniques have been developed for this purpose. Nuclear magnetic resonance, for example, does not require the protein to be available in a crystal form, but is restricted to small proteins. Electron microscopy is used when only 2-D arrays of proteins are available, for example for membrane-bound proteins, which are generally difficult to crystallize. Some 40% of protein structures in a genome are membrane bound.
The most precise technique, however, and the technique capable of tackling the largest molecules is X-ray crystallography. Moreover, around 60% of proteins (being non-membrane bound) are very amenable to crystallography. This technique, which has a long history, has recently been revolutionized by the development of synchrotron radiation sources operating at X-ray wavelengths. Researchers have also made progress in overcoming the two principal bottlenecks in this quest: the growth of high-quality crystals and the well known crystallographic-phase problem.
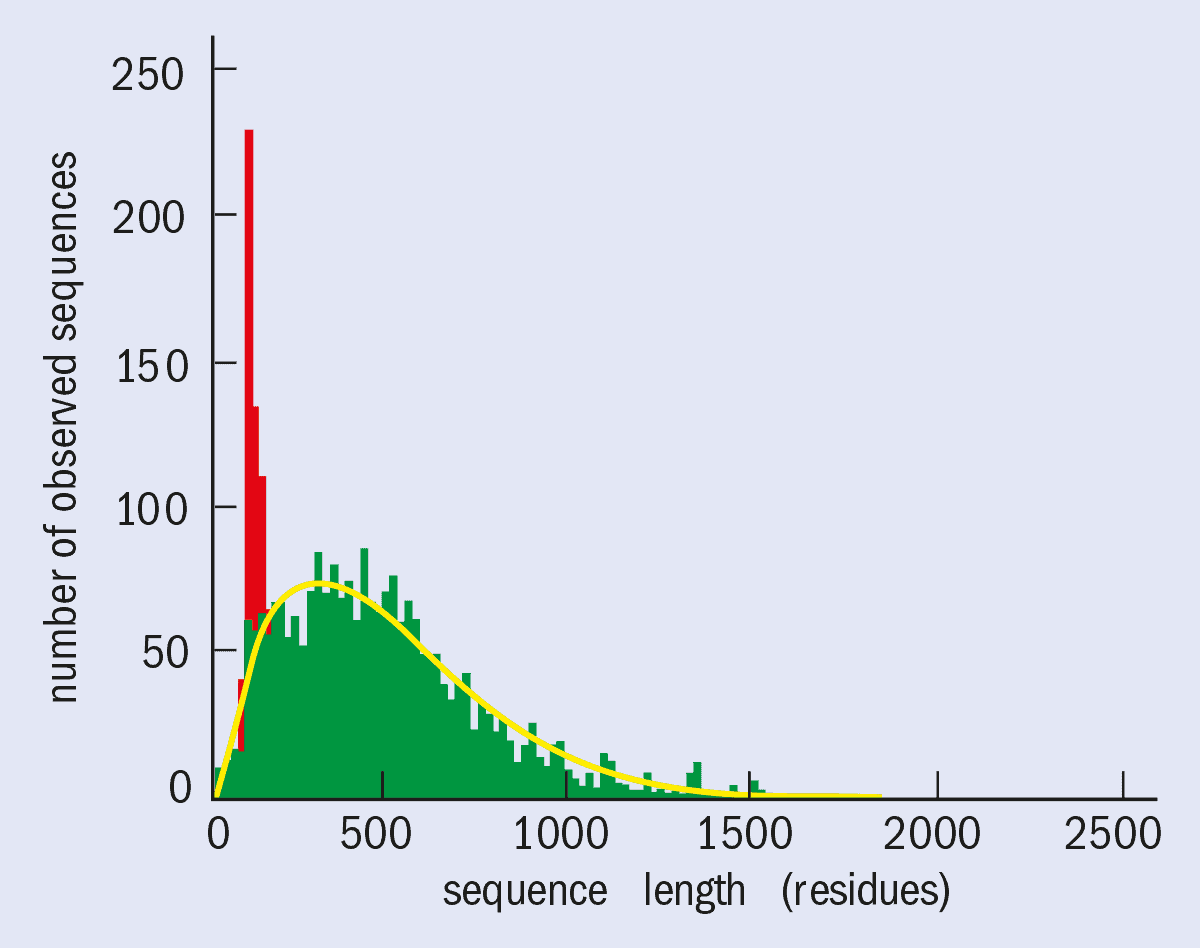
To put the challenge in perspective, it should be noted that there are about 100,000 proteins in the human genome. The yeast genome, which comprises about 10,000 proteins and has already been sequenced, tells us more about the technical challenges. A typical protein contains about 2300 non-hydrogen atoms (i.e. 300 amino-acid residues), but some proteins contain as many as 7000 non-hydrogen atoms (figure 1). The proteins in the human genome are expected to follow a similar distribution.
To date there are some 7000 protein structures in the Protein Data Bank held at the Brookhaven National Laboratory in the US, and this number is doubling approximately every two years, although not all of these structures are “new” proteins since variants (e.g. mutants) of a given protein structure are also deposited. Some 1400 of the 7000 structures are human and 6000 have been determined by X-ray diffraction.
On the basis of current achievements, the determination of the 3-D structures of many proteins in whole genomes has become a realistic prospect. Such a goal is being discussed at the highest levels in synchrotron radiation facilities around the world as part of a possible Genome 3D Structure Determination Project.
Basic crystallography
Protein crystals are quite different to the crystals studied by most physicists. A protein crystal typically has dimensions of ~ 0.5 mm and contains about 1015 protein molecules in a periodic array. However, protein crystallographers are primarily interested in the arrangement of the atoms inside the protein molecule itself, rather than the arrangement of the molecules inside the crystal.
Around half of the protein crystal is actually made of liquid: indeed, a protein crystal becomes disordered if it is allowed to dry out. A small fraction of the solvent binds to the protein to form an ordered shell (or shells), but most of it is found in “bulk solvent channels”. These channels allow the diffusion of smaller molecules or “ligands” into the crystal (e.g. molecules whose reactions are catalysed by enzymes or, just as important, molecules that inhibit enzyme action). These protein-ligand complexes can also be studied by X-ray crystallography, and this sort of work is increasingly being used in the pharmaceutical industry for rational drug design.
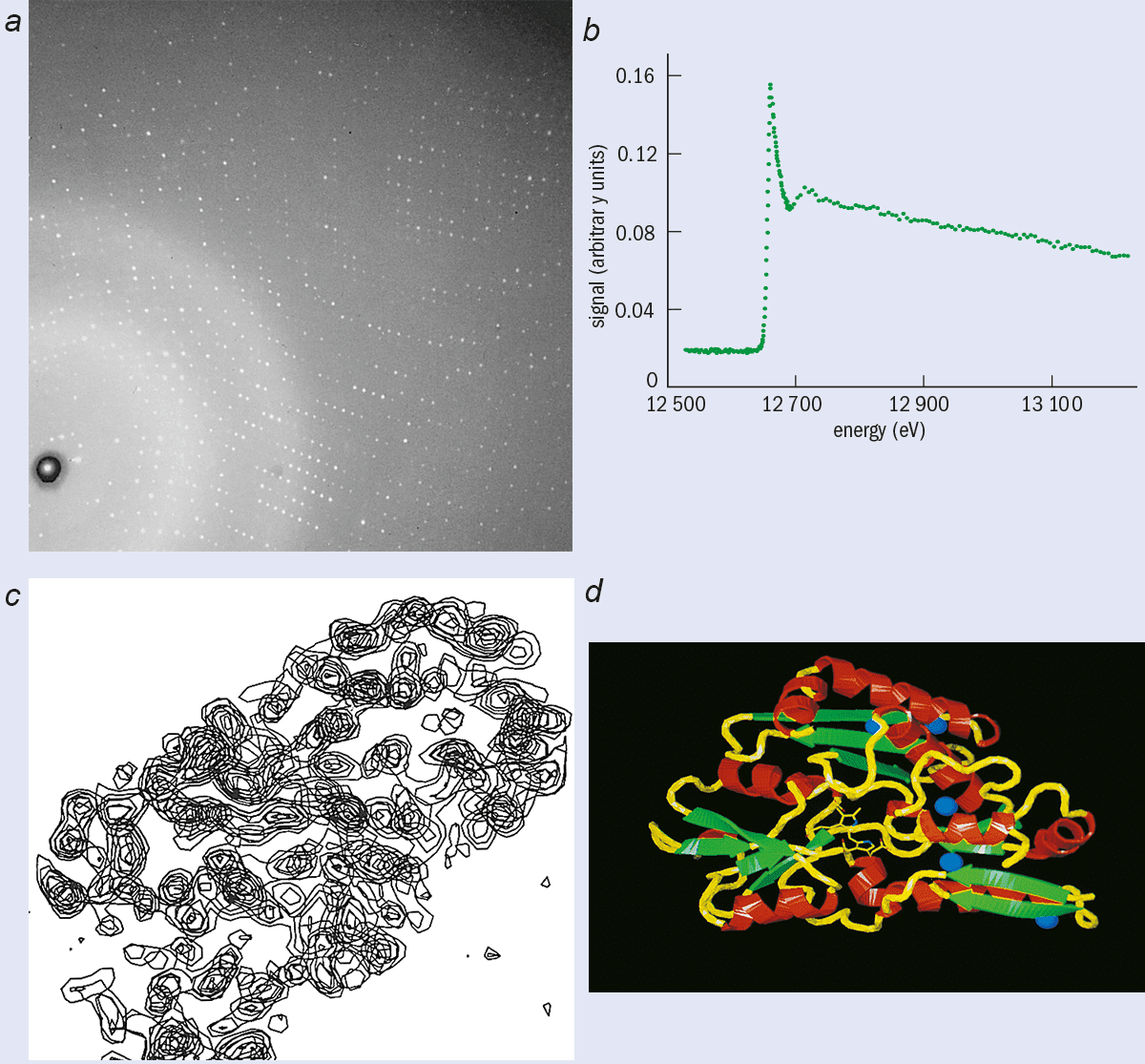
X-ray crystallography relies on the scattering of X-rays by the periodic arrays of atoms and molecules that comprise a single crystal. The scattered X-rays interfere to produce a diffraction pattern that contains a large number of spots in regular positions (see right). Each spot or reflection is governed by Bragg’s law l = 2d sinq, where l is the X-ray wavelength, d is an interplanar spacing in the crystal and 2q is the scattering angle with respect to the incident X-ray beam. For a crystal continuously rotated in an X-ray beam of one particular wavelength, spots light up as the crystal momentarily reflects and then disappear. A complete set of diffraction spots can be measured for a full 360° rotation of the crystal. In practice it is found that the pattern often repeats itself after a smaller rotation (e.g. 90°) due to the internal symmetries of the crystal.
The first crystal structure to be resolved was sodium chloride (NaCl) in 1913, an achievement for which Sir Lawrence Bragg shared the 1915 Nobel Prize for Physics with his father Sir William Henry Bragg. Lawrence Bragg deduced the structure of NaCl by comparing the diffraction patterns of crystals with similar compositions, particularly sodium chloride and potassium chloride (KCl). Although many of the spots were of similar intensity, some of those in the NaCl pattern were missing in the KCl pattern.
Bragg relied on the fact that both crystals were actually diffraction gratings and that the X-rays would therefore undergo constructive or destructive interference from the atomic electrons, producing strong and weak spots, respectively. In particular, Bragg realized that since the potassium and chloride ions both had 18 electrons, destructive interference would lead to missing spots if the crystal contained an alternating array of potassium and chlorine ions in three dimensions. Sodium chloride had a similar structure.
This intuitive approach has been replaced by methods based on Fourier transforms (figure 2). To reconstruct the 3-D arrangement of atoms and molecules responsible for the diffraction pattern with this technique we need to know the amplitude and phase (relative to the incident beam) of each spot in the pattern (see box “Multi-wavelength crystallography”). However, a basic problem is that although the intensity of each spot can be measured, the precise phase cannot. This is a technical rather than fundamental limitation. The problem is that radiation with extremely short wavelengths (~ 1 Å) is needed to probe the interatomic distances (also a few Å) and it is extremely difficult to measure phase at such short wavelengths.
Multi-wavelength crystallography
The electron density, ρ(x,y,z), of the unit cell in a crystal is related to the diffraction data by a Fourier series
where ρ is in units of electron charge per Å3, V is the volume of the unit cell, h, k and l are integers used to label the diffraction spots in three dimensions, |Fhkl| is the amplitude of the hkl-th spot and αhkl is its phase angle. As explained in the text, the big challenge in X-ray crystallography is to determine these phase angles, with a variety of methods having been developed for molecules of different sizes.
The traditional method for doing this for protein crystals has been multiple isomorphous replacement. In this technique, diffraction patterns are collected for the native protein crystal and for two derivative protein crystals in which some atoms have been chemically replaced by heavier atoms. However it can be difficult, and sometimes impossible, to find suitable heavy-atom replacements, since the replacement atom must not disturb the crystal lattice or protein structure. It is also labour intensive.
With modern multi-wavelength anomalous dispersion (MAD) techniques, however, it is possible to introduce a metal atom such as selenium or xenon directly into the protein and to exploit changes in the scattering properties of the atom as a function of synchrotron radiation wavelength. This means that fewer crystals are needed – only one when selenium is used and only two for xenon.
MAD methods rely on the changes to diffraction spots caused by the metal atom. For this to work, the X-ray wavelength must be tuned at the absorption edge of interest in very fine steps, as small as 0.0001 Å. This naturally requires a continuum X-ray source and so has only become possible with the availability of high-brilliance synchrotron radiation sources and improved area detectors. The detectors must be able to efficiently collect tens of thousands of diffraction spots per wavelength with high accuracy.
So how can we replace the sulphur atoms in a protein crystal with selenium? To begin with protein crystals were simply soaked in heavy-atom solutions, but this technique has now been replaced by more reliable “biotech” methods. Human proteins containing selenium can be prepared in bulk by “splicing” human DNA into bacteria and then cultivating the bacteria on a substrate that contains seleno-methionine, an artificial amino acid in which the sulphur has been replaced by selenium. A crystal of the selenium-labelled protein is then grown. The diffraction intensity changes caused by simply changing the incident wavelength to “activate” the selenium atoms can be measured accurately with the modern electron-area detectors; that have replaced photographic film.
Another approach is called single isomorphous replacement with optimized anomalous scattering (SIROAS). In this method, xenon atoms, for example, are forced into pockets on the protein surface by holding the protein crystal at high pressure in xenon gas. Xenon has an X-ray absorption edge at 0.35 Å, so the xenon atoms can be activated by radiation just below this wavelength. In these experiments data are collected at standard pressure – when the protein is in its “native” form and does not contain xenon – and again at high pressure when it does contain xenon. Hence two data sets are measured at one wavelength.
Although the SIROAS signal is much stronger than that from the multi-wavelength technique, the short wavelength of the xenon edge means that only a few synchrotron radiation sources offer the high photon flux needed. Using the 2.27 Å absorption edge in xenon might overcome this problem but this has not been attempted yet. Another possibility is to use iodine, which has absorption edges at 0.33 Å and 2.39 Å. The other disadvantage of the xenon technique is that two crystals are required, unlike the selenium technique that only requires one crystal.
In protein crystallography, the trial-and-error techniques that work with simple compounds are just not practical. Instead a variety of methods have been developed in which heavy atoms with characteristic responses to X-rays are incorporated into the protein crystal. The intensity changes in the diffraction patterns caused by these heavy atoms allow the phases of each and every reflection to be estimated. However it can be difficult, and sometimes impossible, to find suitable heavy atoms that do not disturb the crystal lattice or protein structure. These techniques go under the general name of multiple isomorphous replacement.
One especially productive technique is to replace the sulphur atom in methionine, one of the two amino acids containing sulphur, with selenium (see Hendrickson in further reading). By tuning the wavelength of the synchrotron radiation around the selenium absorption edge at 0.97 Å, it is possible to “activate” the selenium atoms (see previous figure), which leads to intensity changes in the diffraction pattern. It is possible to determine the phase angle of each reflection from the changes in intensity.
Much of the instrumentation and methods for protein crystallography have been pioneered by one of us (JRH) using the Synchrotron Radiation Source at the Daresbury Laboratory in the UK, by Wayne Hendrickson at the National Synchrotron Light Source at Brookhaven and by Roget Fourme at the LURE synchrotron in Paris. More recently these techniques have been extended to the world’s first “third-generation” source, the European Synchrotron Radiation Source (ESRF) in Grenoble, France, by JRH and Andrew Thompson of the European Molecular Biology Laboratory (EMBL), also in Grenoble.
Theorists have also been developing techniques to solve protein structures for over 40 years. However, these techniques could not be properly harnessed until fine-tuning of the X-ray wavelength with synchrotron radiation became possible (see box “What is synchrotron radiation?”).
The combination of synchrotron radiation and multi-wavelength crystallography means that it should now be possible to much more rapidly determine the structure of some 60% of the proteins in the human genome – providing we can grow the crystals.
What is synchrotron radiation?
Synchrotron radiation is produced when a beam of charged particles is bent by a magnetic field. In most sources the beam is confined in a circular or near-circular orbit by magnetic fields and a highly parallel beam of light is emitted. (The energy lost through synchrotron radiation is replaced by radiofrequency electric fields.) The spectrum is continuous and extends into the X-ray region if the beam energy is high enough. Overall, the synchrotron radiation source has many advantages over a laboratory X-ray source. It is more intense, is more collimated and offers a continuum of wavelengths to choose from. In more technical terms, the X-rays have a higher flux (photons per second), a higher brightness (flux per unit solid angle) and a higher brilliance (brightness per unit source area) over this broad spectrum.
In addition to bending magnets, so-called third-generation sources use “insertion devices” – straight sections of magnets – to generate radiation at even shorter wavelengths, and higher brightness and brilliance. Currently the most brilliant source available in Europe is the European Synchrotron Radiation Facility in Grenoble, France – a joint venture between 12 European nations. Similar high-brilliance sources have also recently come on-line in the US and Japan.
The Synchrotron Radiation Source (SRS) at the Daresbury Laboratory, at which much of the work described in this article was carried out, is a second-generation source and is now in urgent need of replacement. The UK synchrotron community is currently trying to secure funding for a proposed replacement source called DIAMOND. Meanwhile two new protein-crystallography beamlines are currently being built at the SRS by Colin Nave and his team. This will give a total of five protein-crystallography beamlines at Daresbury and help build on the lab’s strong track record in this area. This pedigree includes work on the very large multi-protein enzyme (F1-ATPase) that contributed to John Walker of the Laboratory of Molecular Biology in Cambridge sharing the 1997 Nobel Prize for Chemistry.
First grow your crystal
The primary bottleneck in protein crystallography is the production of suitable single crystals. Biocrystallization, like any other crystallization process, involves the classical steps of nucleation and growth, with the molecules having to be brought into a supersaturated, thermodynamically unstable state for crystals to form.
Crystallization of proteins presents a difficult and laborious task because these substances are very sensitive to external conditions. The usual methods of evaporation, high pressure, dramatic temperature variation or the addition of strong organic solvents that are used to grow crystals of semiconductors, superconductors, diamonds and so on, simply do not work for proteins. Gentler techniques such as diffusion, dialysis and batch crystallization are needed (see box “Protein crystal growth”). All of these techniques aim to guide the protein gently out of the solution and into a crystal.
There has never been a set rules or recipes that explain how to crystallize a new protein. Indeed, there is generally no indication that one is close to crystallization conditions until a crystalline precipitate or the first crystals appear. Efficient methods are therefore required to help the experimenter to find a lead, which allows the crystallization conditions to be optimized. Crystallization therefore breaks down into two stages: “screening”, in which various different experimental conditions are tried to obtain crystals of any description; and optimization, where one tries to improve the size and quality of the crystals.
Although the idea of screening has been around since the late 1970s, it did not become popular because it was, basically, laborious, time consuming and boring. However, since the development of automation, screening has become much more widely used, and this has significantly raised the success rate of obtaining suitable crystals of a wide range of proteins.
However, the vast number of experiments that have to be performed to hit upon the correct range of conditions consume considerable amounts of material, and many of the more interesting proteins are only available in limited supply. On average 5 mg of pure protein is needed for screening but sometimes only 1 or 2 mg is available. The efficiency of the search can be improved by statistical means, which can help to minimize the amount of protein used, but there is still demand for techniques that rapidly obtain as much information as possible on a protein while using minimal amounts of material.
The amount of material needed can be reduced by using smaller volumes of protein solution, but evaporation sometimes causes the sample to dry out before it crystallizes. The problem of evaporation can be overcome by dispensing and incubating the samples under oil (paraffin oil, silicone oils and combinations of these). Small drops containing only 1-2 microlitres of a mixture of the protein and various crystallizing agents are dispensed through a very fine tip into the oil, where they are protected from evaporation. This approach, developed by a collaboration between Imperial College and Douglas Instruments, both in London, has resulted in enormous savings of protein.
The oil has other benefits in addition to preventing evaporation. For example, it protects the samples from airborne contamination (which can cause excess unwanted nucleation). This also enhances the cleanliness of the trials, thereby leading to more accurate and reproducible experiments. Furthermore, once the crystals have formed they are buoyed up by the viscous oil. This makes the crystals resistant to physical shock and easier to transport (e.g. to and from synchrotron radiation sources).
Despite the success of screening it is clear that even the most successful crystallization methods still rely on trial and error rather than on an analytical approach. However, by monitoring crystallization with diagnostic instruments it should be possible to understand and optimize the growth process. In the ideal case, the researcher would be able to intervene in the crystallization process as it proceeds, allowing the experiment to be steered to the desired result. For example, nucleation and growth require different conditions and so the outcome could be greatly improved if the conditions were changed during the experiment to reflect this.
A wide variety of techniques is available to monitor nucleation and growth, notably light scattering and interferometry, and our understanding of the best conditions for growth is increasing. Although such diagnostic experiments are time consuming and, like screening, consume a lot of material, they are of major importance and are performed in parallel with the current empirical methods.
Protein crystal growth
There are four major techniques used to grow protein crystals: batch crystallization, vapour diffusion, liquid-liquid diffusion and dialysis. The typical time for protein crystal growth is 1 to 3 weeks, although timescan vary from several hours to a year. Batch crystallization is the oldest and simplest method: the protein to be crystallized is mixed with the crystallizing agents at the required concentrations at the start of the experiment and left to crystallize.
The various diffusion/dialysis methods are dynamic systems that involve attaining an equilibrium between an aqueous drop containing the protein solution and a reservoir containing the crystallizing agents (i.e. buffer, precipitants, additives). A gradual change of the conditions takes place in the drop en route to equilibrium, often resulting in the formation of crystals.
No one method is considered superior to another. The choice of method depends on the protein involved, its quantity and mainly on the preference of the experimenter. Vapour diffusion and batch are the most widely
used techniques mainly due to the ease with which they can be set up in comparison with the other methods. Moreover, automation has been developed for both.
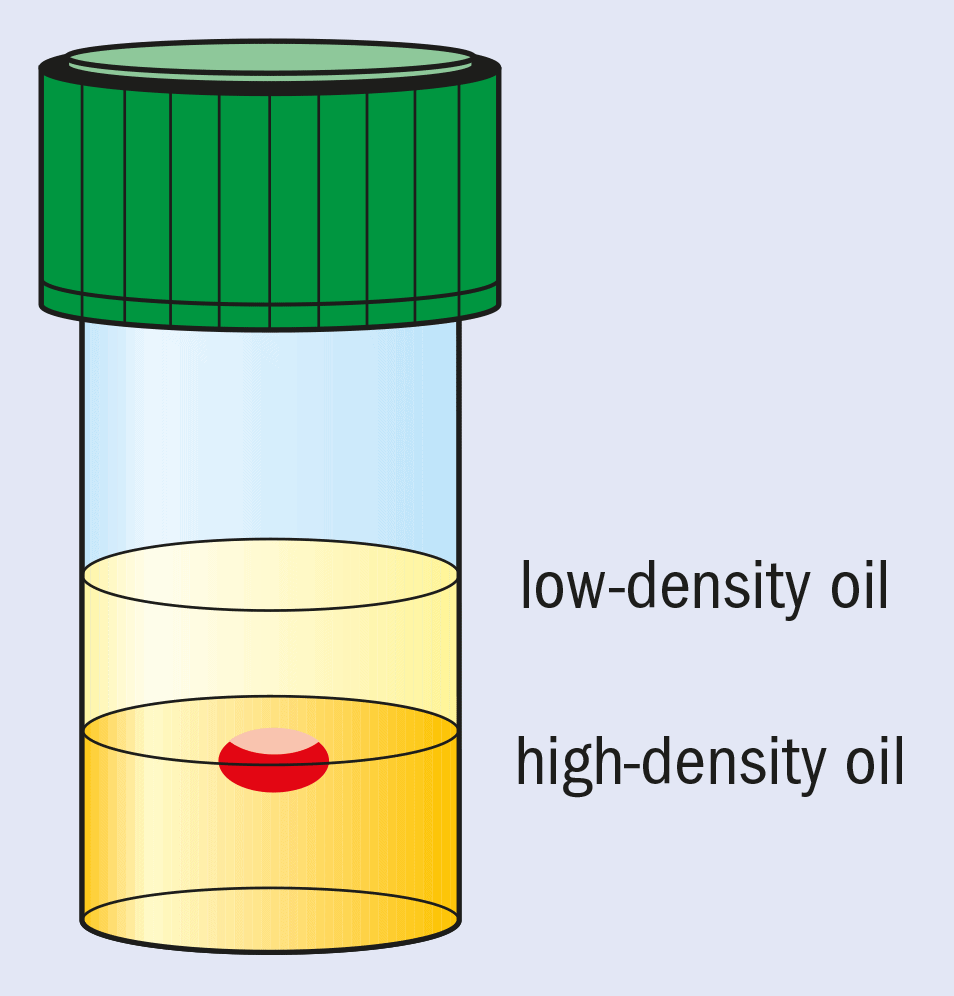
Another approach is to employ microgravity conditions to eliminate convection and sedimentation. This allows solely diffusion-driven conditions to be established. These are thought to be ideal for high-quality crystal growth and, in particular, the growth of crystals with weak intermolecular forces and lattice interactions. Microgravity conditions for crystallization can be partially mimicked by growing crystals in oil.
This method, pioneered at Imperial College, involves “containerless” growth with the drop containing the protein and the crystallizing agents suspended between two different oils with carefully chosen densities (see figure). The bottom layer is a high-density (1.27 g cm–3) fluorinated silicone fluid and the top layer contains low-density standard silicone fluid (0.92 g cm–3).
Experimental challenges
The actual X-ray data collection, if performed at or near room temperature, can lead to serious radiation damage to the crystals due to their high water content. The X-rays generate free radicals, which travel through the solvent channels that criss-cross the crystal and attack the intermolecular contacts between the adjacent protein molecules. This eventually leads to the break up of the crystal. However, the channels can also be used to diffuse small molecules to the active sites on the protein. This forms the basis of the growing field of time-resolved macromolecular crystallography.
High-speed data collection and short wavelengths do reduce damage significantly but the best approach is to freeze the crystal by plunging it into liquid nitrogen or propane. It might be thought that the expansion of the water when it freezes into ice would damage the crystal, but this can be avoided if the temperature is reduced quickly enough. Elspeth Garman of the molecular biophysics department at Oxford University in the UK is a pioneer of these techniques, while Mike Glazer of the physics department at Oxford has developed liquid-nitrogen apparatus that is economic to run and that is now marketed by Oxford Cryosystems.
So far the best results have been obtained with very small crystals (less than 300 microns). However, this is not a problem because the high brilliance of synchrotron X-ray beams still allows excellent data to be collected from such small crystals.
Freezing changes the protein structure slightly and it is obviously important to take this into account when using such structures in the design of drugs that will have to work at room temperature. However, this slight complication is greatly outweighed by the reduction in radiation damage to the crystal sample.
Position-sensitive X-ray detectors are another area of active research. In the UK, for example, Oxford Instruments is leading the multi-million pound IMPACT (Innovative Microelectronic Pixellated Sensors and Advanced CCD Technology) programme to develop novel X-ray detectors that combine charge coupled devices and pixellated silicon detectors. The aim of IMPACT, which involves some of the UK’s most successful instrumentation companies working with the academic research community, is to develop new detectors for both commercial and scientific applications. Other efforts are centred at the CHESS synchrotron at Cornell University in the US, the University of California at San Diego and the Advanced Light Source at the Lawrence Berkeley National Laboratory, also in California.
The combination of a frozen protein crystal that is radiation hard, an extremely intense X-ray beam and a sensitive area detector such as a charge-coupled device (CCD) has opened up a new territory in determining protein structures. In Manchester, for example, we have worked closely with researchers at the CHESS synchrotron, along with structural biologists from the Weizmann Institute in Israel, to determine the structure of concanavalin A, a 25 000 molecular weight protein isolated from plants, at a resolution of 0.94 Å. Much research and development in this area has been conducted at the EMBL Outstation in Hamburg by Keith Wilson and Zbyszek Dauter.
These experiments also allow the structure and bonding details of many of the hydrogen atoms in the protein to be seen directly, even those bound in water molecules, traditionally a speciality of neutron protein crystallography. However, if we want to study the exchange of these hydrogen atoms with an aqueous medium – which is relevant in catalysis, for example – we need to use neutrons. At one time it was thought that proteins like concanavalin A were just too large to be studied with neutron protein crystallography. However, a pilot experiment at the Institut Laue-Langevin neutron source in Grenoble has shown that it is possible to collect enough data to study the proton exchange process if a range of neutron wavelengths and a large-area image plate detector are used. The experiment also demonstrates the wide range of techniques that are sometimes needed to understand the function of just one protein.
Proteins are also of considerable industrial and medical interest. For example, concanavalin A binds glucose, and the structure of this complex, also determined in Manchester, now forms the basis of a glucose-based biosensor for diabetics that is being developed by Hugh Jones of the University of Wales Swansea and industry. Similar plant proteins, known as lectins, might be able to prevent HIV infection by competing with the HIV virus as it tries to attach itself to cells. This approach is being investigated by Pierre Rizkallah of Daresbury and Colin Reynolds of Liverpool John Moores University in the UK.
The future
There has been remarkable progress in protein crystallography in recent years. Indeed the determination of all of the protein structures in the human genome is now a realistic prospect. How long might this take? If we assume no further breakthroughs in techniques, it takes one or two days of beam-time at a synchrotron radiation source to acquire the diffraction data that will allow a typical protein structure to be determined. Twenty instruments working world-wide in a coordinated way would then require about 10,000 days or almost 30 years of work.
However, improved position-sensitive X-ray detectors could speed up this process enormously. For example, the pixel detectors would yield data sets for each of two wavelengths or crystals in matters of minutes, rather than hours or days.
Therefore, the determination of genome level structure determination might well take considerably less than 30 years, assuming that we can grow the crystals fast enough. It would have vastly important consequences for improved pharmaceutical design and the understanding of genetic diseases. Overall this would be physics, with chemistry and biophysics, applied at its best.



