The study of protein folding is fundamental in biophysics because it is important for understanding how proteins function in a wide range of biological processes and for investigating diseases like Alzheimer’s and Parkinson’s in which proteins misfold. Thanks to a new quantum approach, researchers in China have now found that proteins could fold much faster than previous calculations suggest.
Proteins consist of a long chain of molecules known as amino acids folded into a 3D shape. Researchers have been studying protein folding since the 1950s, and in 1956, two postdocs working at the Carlsberg Laboratory in Copenhagen, John Schellman and Bill Harrington, were the first to discover that protein folding reactions are very fast and often reversible. However, just over a decade later, physicist and molecular biologist Cyrus Levinthal pointed out that they may be extremely slow.
The Levinthal paradox
In his model, Levinthal assumed that a protein folds through a series of meta-stable intermediate states and that a myriad of different conformations are possible. He estimated that if a protein explored new conformations randomly at the rate at which a single molecular bond can rotate, the time it would take for it to explore all the possible conformations would amount to the age of the universe – even for a protein containing just 100 amino acid residues. Experiments show, however, that proteins can fold in less than a few seconds.

There are many theoretical models today to explain this so-called Levinthal paradox. These include the Ising-like model, the foldon-dependent protein folding model and the nucleation-condensation mechanism. All these models require that simple assumptions of the protein structure be made from the outset, however.
A quantum walk on a definite graph
Researchers at Zhejiang University in Hangzhou led by You-Quan Li and Li-Hua Lu have now put forward a quantum strategy to describe protein folding as a quantum walk on a definite graph. This, they say, provides them with a general scheme without having to resort to simple, “artificial”, hypotheses.
“In our technique, we calculate the folding time as the mean first-passage time from the protein’s initial state (a straight-line conformation) to the target, final state (the most compact structure that models the native conformation),” explains Li.

Protein folding time is much shorter
The protein folding time Li and Lu obtained is much shorter than that calculated from classical random walks. “This result should substantially advance the research field of protein folding,” says Li. “It may also help in the development of protein engineering technologies and in the design of protein-based nanodevices.”
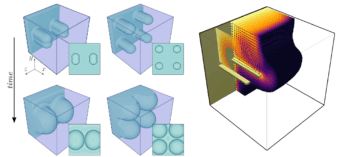
In their work, they describe the protein structure by the frequently adopted 2D square lattice model in which a protein is thought of as a chain of non-intersecting units (amino-acid residues). The chains have a given length on the lattice.
“For a protein with n-amino-acid residues, we can calculate the total number Nn of distinct lattice conformations that are different for each protein intermediate structure,” explains Lu. “For example, we have N4= 4 and N6= 22. This technique provides us with a set of Nn objects (a structure set).”
One-step folding process
The researchers say they studied the protein folding process as a one-step folding. “Based on the lattice model, we can naturally define this one-step folding by the displacement of one amino acid in one of the lattice sites,” says Lu. “This approach allows us to establish certain connections between distinct points in a structure set and to draw up a connection graph.
Amino acid orchestra trains machine learning algorithms to design proteins
“In other words, two structures are connected via one-step folding if their conformations differ in one site only.”
The model, as it stands, does have its limitations though, insists Li. “In this work, we have put forward a self-contained quantum approach to investigate the problem of protein folding without artificial hypotheses, but we have only considered the case of six residues as a model study.
“For a genuine interpretation of experimental protein-folding time, we will need to apply our approach to more residues,” he tells Physics World.
The research is detailed in Chinese Physics Letters 10.1088/0256-307X/36/8/080305.