Combining physics, biology and chemistry, structural biology investigates the anatomy of biological macromolecules, and proteins in particular, improving understanding of diseases and enabling drug discovery. Speaking at the 68th Lindau Nobel Laureate meeting in Germany last week, biophysicist Joachim Frank says that the field is entering a new era with a bright future thanks to advances in cryogenic electron microscopy (cryo-EM).
Frank, a German-American Nobel Laureate in chemistry based at Columbia University, New York, traced the history of cryo-EM to the present day in his Lindau lecture. A technique pioneer, Frank shared the 2017 prize with fellow biophysicists Richard Henderson and Jacques Dubochet, for the significant contributions he made to its advancement.
Cryo-EM rapidly freezes molecular samples in solution and images them with an electron beam to reveal the molecular structure. Today, resolutions of 3-4 Å are routinely achievable, while at its upper limits, the technique can achieve atomic resolutions of 2 Å.
The technique’s particular strength is its ability to construct models of single, unattached molecules from images of the molecules in their natural state – encompassing a range of arrangements and binding states with other molecules.
A mainstay for structure determination, X-ray crystallography, in contrast, demands molecules are prepared as crystals, a form not typically taken by biomolecules. Crystallography can, however, take advantage of the regularly-ordered molecules to obtain diffraction patterns with which their structures can be reconstructed.
Physicists bag chemistry Nobel for cryo-electron microscopy

In the early 1970s, researchers saw electron tomography of individual molecule as a solution. However, in a major drawback, images of intact molecules were not possible, as the doses required inflict significant damage on the samples.
At around the same time, during his PhD, Frank had a new idea. It was to use computational and mathematical techniques to wrangle 2D images of hundreds of thousands of molecules in a sample. By aligning and averaging them, a single, clearer 3D picture of the molecule could be constructed, he proposed. The ribosome, a molecular machine found in large quantities in the cell cytosol that makes proteins, was to become his test object in developing the techniques.
A workbench for molecular reconstruction
To process and reconstruct the cryo-EM images, Frank developed SPIDER, a modular image processing program and the first of its kind in electron microscopy, around the turn of the 1980s. The computational equivalent of a workbench, it comprised hundreds of operations.
Key techniques developed in Frank’s lab included a correlation averaging technique to identify and average ribosomes facing in a particular direction into a single structure. In another, multivariate statistical analysis was applied to tackle the structural heterogeneity that occurs across a population of molecules in a sample, classifying and grouping similar structures together. Bringing all the techniques together, Frank and post-doc Michael Radermacher completed their first molecular reconstruction, the large subunit of the ribosome, in 1986.
Around the same time, Dubochet and colleagues at the European Molecular Biology Laboratory in Heidelberg made an important advance. For the first time, Dubochet vitrified sample solutions into a glass-like fluid by rapid freezing using liquid ethane cooled to -196°C. The technique prevents the formation of ice crystals that otherwise damage the molecules and diffract the electron beam, resulting in unusable images.
“This now gave the method I developed a very big boost, because now we could look at molecules in their native states,” said Frank. Exploiting this, Frank and collaborators went on to reconstruct several structures for the first time in the mid-1990s. They included a calcium release channel, octopus haemocyanin and the Escherichia coli ribosome. “These were all pioneering contributions at the time.”
Revealing ribosome movement
Another landmark finding followed in 2000. Frank and post-doc Rajendra Agrawal took advantage of further improvements in resolution to reveal the structure of the Escherichia coli ribosome in unprecedented detail. Their analysis revealed a ratchet-like movement of the ribosomes’ two subunits relative to one another during translation, the process where messenger RNA is used synthesize proteins. The movement proved critical to the ribosome’s functioning.
Despite the breakthroughs, however, Frank still saw resolution as a limiting factor in the lab’s research. In 2013, they achieved their best imaging on film after two years going through 260,000 images of the ribosome of Trypanosoma brucei, a parasite that causes African sleeping sickness. “We got stuck at 5.5 Å resolution. It was essentially a wall.” At this resolution, it was not possible to infer structures with high precision. Side chains on the molecules, for example, require a resolution of around 3 Å.
Cameras take cryo-EM to next level
Arrival of the first commercial single electron detection cameras in 2012, however, has had a decisive impact. Their detective quantum efficiency (DQE) is significantly higher than that of film. Frank’s lab used them in 2016 to resolve the structure of the ribosome in Trypanosoma cruzi, a parasite responsible for Chagas disease, at a resolution of 2.5 Å. His technique, with the aid of the camera even allowed a single water molecule to be resolved. “I couldn’t believe it when I first saw it,” said Frank.
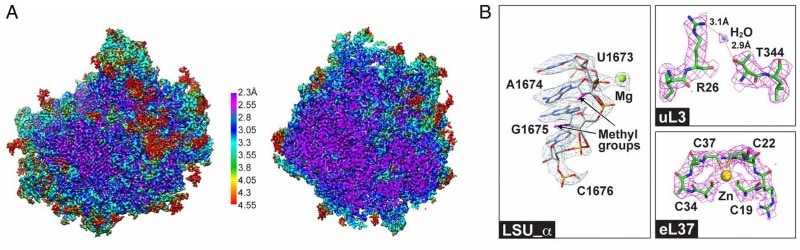
Combined with maximum likelihood statistical methods, cryo-EM imaging technology is now enabling the determination of multiple, co-existing structures in a given sample at near-atomic resolution. This often results in snapshots of molecules in different states. Dubbed a “story in a sample” by Frank, the information means cellular functions can be visualized indirectly. Through these capabilities, he predicts a boom in knowledge. “There’s going to be a huge expansion of [structural] databases relevant for molecular medicine.”
Joachim Frank’s full lecture, Single-Particle Cryo-EM of Biological Molecules – the Sky Is the Limit, can be seen below. (Courtesy: Lindau Nobel Laureate meetings)